 Bệnh viện đa khoa tỉnh Quảng Trị | Bệnh viện tỉnh Quảng Trị | BVĐK Quảng Trị
Bệnh viện đa khoa tỉnh Quảng Trị | Bệnh viện tỉnh Quảng Trị | BVĐK Quảng Trị

Thalassemia thực trạng và giải pháp
Thalassemia thực trạng và giải pháp
1. Tổng quan Thalassemia, chẩn đoán và điều trị
Bệnh Thalassemia là nhóm bệnh thiếu máu di truyền lặn do đột biến gen tổng hợp globin làm giảm hoặc không sản xuất globin để tạo thành hemoglobin (Hb), gây ra tình trạng thiếu máu. Bệnh có 2 nhóm chính là α-thalassemia và β-thalassemia tùy theo nguyên nhân đột biến ở gen α-globin hay β-globin. Đây là bệnh thiếu máu di truyền phổ biến trên thế giới, phân bố khắp toàn cầu nhưng có tính địa dư rõ rệt: tỷ lệ cao ở Địa Trung Hải, Trung Đông, Châu Á, Thái Bình Dương.
Bệnh Thalassemia là nhóm bệnh thiếu máu di truyền lặn do đột biến gen tổng hợp globin làm giảm hoặc không sản xuất globin để tạo thành hemoglobin (Hb), gây ra tình trạng thiếu máu. Bệnh có 2 nhóm chính là α-thalassemia và β-thalassemia tùy theo nguyên nhân đột biến ở gen α-globin hay β-globin. Đây là bệnh thiếu máu di truyền phổ biến trên thế giới, phân bố khắp toàn cầu nhưng có tính địa dư rõ rệt: tỷ lệ cao ở Địa Trung Hải, Trung Đông, Châu Á, Thái Bình Dương.
Bệnh α-thalassemia xuất hiện ở tất cả các khu vực, các quốc gia cũng như các chủng tộc trên thế giới, với khoảng 5% dân số thế giới mang gen bệnh. Thể bệnh lâm sàng nặng nhất của bệnh alpha thalassemia là phù thai Hb Bart’s. Người phụ nữ có thai bị phù thai Hb Bart’s là một trường hợp thai nghén có nguy cơ cao cả về phía mẹ và về phía thai. Về phía thai: thường thai chết trong tử cung hoặc ngay sau sinh. Về phía mẹ: nếu có kèm phù rau thai thì mẹ nhiều nguy cơ tiền sản giật và băng huyết sau đẻ.
Bệnh β-thalassemia thường thấy ở người gốc Trung Đông, Địa Trung Hải, Ấn Độ và Đông Nam Á. Thể bệnh lâm sàng nặng nhất của bệnh beta thalassemia với kiểu gen bệnh đồng hợp tử, có biểu hiện bệnh thiếu máu tan máu nặng nề với nhiều biến chứng trên nhiều cơ quan của cơ thể. Trẻ bị β-thalassemia đồng hợp tử khi sinh ra vẫn mạnh khỏe nhưng sẽ có các triệu chứng bệnh lý thalassemia thể nặng sớm từ ngay trong năm đầu đời. Những người bệnh này cần điều trị truyền máu và thải sắt suốt đời và chất lượng cuộc sống thấp do các biến chứng của bệnh.
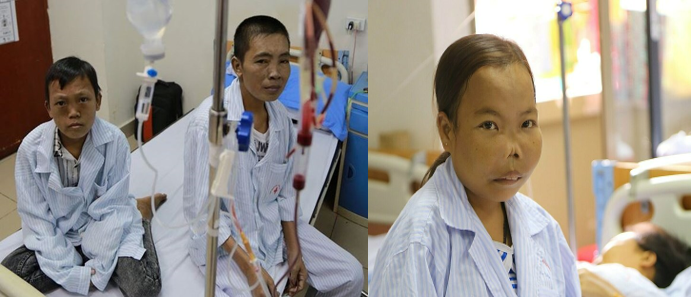
Chẩn đoán bệnh dựa vào:
- Tiền sử: gia đình có người bị bệnh thalassemia.
- Triệu chứng lâm sàng: thiếu máu, vàng da, lách to, biến dạng xương, xạm da,...
- Triệu chứng cận lâm sàng:
+ Tổng phân tích tế bào máu ngoại vi: thiếu máu (Hemoglobin giảm), hồng cầu nhỏ (MCV giảm), hồng cầu nhược sắc (MCH giảm), kích thước hồng cầu không đều (RDW tăng), hồng cầu non ra máu ngoại vi, số lượng hồng cầu lưới tăng.
+ Sức bền hồng cầu tăng.
+ Sinh hóa: bilirubin toàn phần tăng, bilirubin gián tiếp tăng, sắt huyết thanh tăng, ferritin tăng,...
+ Điện di huyết sắc tố: có và/hoặc tăng tỷ lệ huyết sắc tố bất thường: HbA2, HbF, HbE, HbH, HbBart’s, HbCs, HbQs, HbS, HbC,...
+ Di truyền phân tử: các xét nghiệm di truyền học phân tử như Multiplex PCR, giải trình tự gen,… để phân tích các đột biến gen trên các gen α và β globin.
Điều trị:
- Điều trị chính cho người bệnh thalassemia là truyền máu và thải sắt.
+ Truyền máu đều đặn suốt đời đóng vai trò trọng tâm trong điều trị bệnh thalassemia đặc biệt cho người bệnh thalassemia thể nặng.
+ Người bệnh thalassemia thể nặng dư thừa sắt trong cơ thể, sự dư thừa này sẽ làm tích tụ sắt ở các cơ quan trong cơ thể gây tổn thương các cơ quan ấy, đặc biệt là tim, gan gây hậu quả xơ gan, suy tim. Cơ thể không có cơ chế thải một lượng sắt dư thừa quá mức do đó người bệnh thalassemia phải được dùng các thuốc thải sắt.
- Bên cạnh đó, một số biện pháp khác cũng được áp dụng trong điều trị thalassemia như cắt lách, ghép tế bào gốc tạo máu, liệu pháp gen, thụ tinh trong ống nghiệm với những phôi không bị mang gen,...
- Chăm sóc toàn diện: Để phòng ngừa và hạn chế các biến chứng, nâng cao chất lượng cuộc sống cho người bệnh.
- Điều trị biến chứng: Tùy theo biểu hiện, điều trị biến chứng như suy tuyến nội tiết, đái tháo đường, suy tim, xơ gan, loãng xương, rối loạn đông máu,...
- Tiền sử: gia đình có người bị bệnh thalassemia.
- Triệu chứng lâm sàng: thiếu máu, vàng da, lách to, biến dạng xương, xạm da,...
- Triệu chứng cận lâm sàng:
+ Tổng phân tích tế bào máu ngoại vi: thiếu máu (Hemoglobin giảm), hồng cầu nhỏ (MCV giảm), hồng cầu nhược sắc (MCH giảm), kích thước hồng cầu không đều (RDW tăng), hồng cầu non ra máu ngoại vi, số lượng hồng cầu lưới tăng.
+ Sức bền hồng cầu tăng.
+ Sinh hóa: bilirubin toàn phần tăng, bilirubin gián tiếp tăng, sắt huyết thanh tăng, ferritin tăng,...
+ Điện di huyết sắc tố: có và/hoặc tăng tỷ lệ huyết sắc tố bất thường: HbA2, HbF, HbE, HbH, HbBart’s, HbCs, HbQs, HbS, HbC,...
+ Di truyền phân tử: các xét nghiệm di truyền học phân tử như Multiplex PCR, giải trình tự gen,… để phân tích các đột biến gen trên các gen α và β globin.
Điều trị:
- Điều trị chính cho người bệnh thalassemia là truyền máu và thải sắt.
+ Truyền máu đều đặn suốt đời đóng vai trò trọng tâm trong điều trị bệnh thalassemia đặc biệt cho người bệnh thalassemia thể nặng.
+ Người bệnh thalassemia thể nặng dư thừa sắt trong cơ thể, sự dư thừa này sẽ làm tích tụ sắt ở các cơ quan trong cơ thể gây tổn thương các cơ quan ấy, đặc biệt là tim, gan gây hậu quả xơ gan, suy tim. Cơ thể không có cơ chế thải một lượng sắt dư thừa quá mức do đó người bệnh thalassemia phải được dùng các thuốc thải sắt.
- Bên cạnh đó, một số biện pháp khác cũng được áp dụng trong điều trị thalassemia như cắt lách, ghép tế bào gốc tạo máu, liệu pháp gen, thụ tinh trong ống nghiệm với những phôi không bị mang gen,...
- Chăm sóc toàn diện: Để phòng ngừa và hạn chế các biến chứng, nâng cao chất lượng cuộc sống cho người bệnh.
- Điều trị biến chứng: Tùy theo biểu hiện, điều trị biến chứng như suy tuyến nội tiết, đái tháo đường, suy tim, xơ gan, loãng xương, rối loạn đông máu,...
2. Thực trạng thalassemia trên thế giới và Việt Nam
Theo báo cáo của Liên đoàn Thalassemia thế giới năm 2013, có khoảng 7% dân số trên thế giới mang gen bệnh huyết sắc tố. Theo số liệu thống kê của WHO-2008, bệnh huyết sắc tố ảnh hưởng tới 71% các nước trên thế giới. Mỗi năm có khoảng 60.000-70.000 trẻ em sinh ra bị bệnh β-halassemia mức độ nặng. Bệnh huyết sắc tố là nguyên nhân gây ra 3,4% các trường hợp tử vong ở trẻ em dưới 5 tuổi. Trên toàn thế giới có khoảng 7% phụ nữ mang thai có gen bệnh huyết sắc tố và khoảng 1,1% các cặp vợ chồng có nguy cơ sinh con bị bệnh. Có hơn 200 quốc gia và vùng lãnh thổ chịu ảnh hưởng bởi căn bệnh này trong đó có Việt Nam.
Theo báo cáo của Liên đoàn Thalassemia thế giới năm 2013, có khoảng 7% dân số trên thế giới mang gen bệnh huyết sắc tố. Theo số liệu thống kê của WHO-2008, bệnh huyết sắc tố ảnh hưởng tới 71% các nước trên thế giới. Mỗi năm có khoảng 60.000-70.000 trẻ em sinh ra bị bệnh β-halassemia mức độ nặng. Bệnh huyết sắc tố là nguyên nhân gây ra 3,4% các trường hợp tử vong ở trẻ em dưới 5 tuổi. Trên toàn thế giới có khoảng 7% phụ nữ mang thai có gen bệnh huyết sắc tố và khoảng 1,1% các cặp vợ chồng có nguy cơ sinh con bị bệnh. Có hơn 200 quốc gia và vùng lãnh thổ chịu ảnh hưởng bởi căn bệnh này trong đó có Việt Nam.
Theo Viện Huyết học – Truyền máu Trung ương, tại Việt Nam tất cả 63 tỉnh và 54 dân tộc đều có người mang gen bệnh, với tỷ lệ mang gen bệnh trên 13% thì ước tính có khoảng 14 triệu người mang gen bệnh trên cả nước, nhiều dân tộc tỷ lệ mang gen thalassemia lên tới 30-40%, riêng dân tộc Kinh là 9,8%. Các dân tộc ở khu vực Bắc bộ và Bắc trung bộ có tỷ lệ mang gen α0 thalassemia và β0 thalassemia cao, dân tộc sinh sống ở Tây Nguyên, Nam Trung Bộ và Nam Bộ có tỷ lệ mang gen α+ thalassemia và HbE cao.
Mỗi năm, ước tính cả nước có thêm khoảng 8.000 trẻ em sinh ra bị bệnh thalassemia, trong đó có khoảng 2.000 trẻ bị bệnh mức độ nặng và khoảng 800 trẻ không thể ra đời do phù thai. Một người bệnh mức độ nặng từ khi sinh ra đến năm 30 tuổi cần khoảng 3 tỷ đồng để điều trị và đến năm 21 tuổi cần truyền khoảng 470 đơn vị máu để duy trì đời sống. Với trên 20.000 người bệnh mức độ nặng cần phải điều trị cả đời. Việt Nam cần có trên 2.000 tỷ đồng mỗi năm để cho tất cả bệnh nhân có thể được điều trị tối thiểu và cần có khoảng 500.000 đơn vị máu an toàn.

Trên địa bàn tỉnh Quảng Trị, có dân tộc Bru Vân Kiều nằm trong nhóm các dân tộc có tỷ lệ mang gen α+ thalassemia cao (62%) và tỷ lệ HbE cao (52,7%). Ước tính tỷ lệ trẻ sinh ra bị bệnh thalassemia trong 1000 ca sinh của tỉnh Quảng Trị lần lượt:
+ Tỷ lệ Hb Bart: 0,25%.
+ Tỷ lệ HbH: 0,79%.
+ Tỷ lệ Beta thalassemia và Beta thalassemia/HbE: 0,14%.
Theo một nghiên cứu về thalassemia được thực hiện tại bệnh viện đa khoa tỉnh Quảng Trị năm 2020 cho kết quả như sau: Bệnh nhân chủ yếu là đối tượng trẻ dưới 20 tuổi (54,9%) và không có sự khác biệt nhiều về giới, bệnh gặp ở cả người Kinh và ngươi Bru Vân Kiều, bệnh nhân vào viện chủ yếu vì tình trạng thiếu máu (62.7%) với biểu hiện da, niêm mạc nhợt nhạt (90,2%), gặp nhiều nhất là β thalasemia (72.5%), ít gặp hơn là α thalasemia (27.5%), mức độ thiếu máu trung bình đến nặng chiếm đa số (68,7%), quá tải sắt (45,1%), tổng đơn vị máu truyền trong năm 282 đơn vị/ 51 bệnh, trung bình 5.5 đơn vị /bệnh /năm.
3. Một số giải pháp
Theo kinh nghiệm của các nước trên thế giới, thalassemia là bệnh có thể chữa được, phòng được, tiến tới chấm dứt tình trạng phù thai do Thalassemia, tình trạng trẻ sinh ra bị bệnh và giảm dần tỷ lệ di truyền gen bệnh trong cộng đồng. Mặt khác, qua thực tiễn cho thấy, chi phí đầu tư cho phòng bệnh nhỏ hơn rất nhiều so với chi phí điều trị và hiệu quả mang lại rất cao.
Có ba giải pháp chính sau:
- Thứ nhất là chẩn đoán và điều trị sớm bệnh thalassemia giúp nâng cao chất lượng cuộc sống của người bệnh.
- Thứ hai là tư vấn tiền hôn nhân, giúp cho người dân biết mình có mang gen bệnh không và có nguy cơ sinh con bị bệnh thalassemia thể nặng không, nhưng không ngăn cản được việc kết hôn.
- Thứ ba là sàng lọc và chẩn đoán trước sinh, giúp chẩn đoán sớm thai bị bệnh thalassemia thể nặng ở tuổi thai nhỏ để tư vấn cho gia đình có thể ngừng thai nghén, giúp cho gia đình và xã hội giảm những gánh nặng chăm sóc và điều trị những người bệnh thalassemia thể nặng.
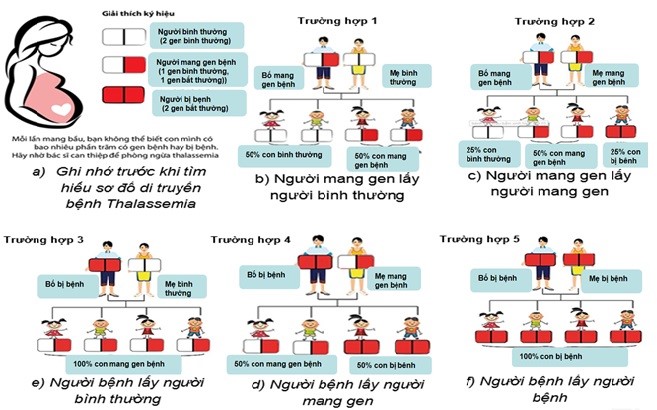
Trong đó tư vấn tiền hôn nhân, sàng lọc và chẩn đoán trước sinh đóng vai trò quan trọng trong việc dự phòng ngay từ đầu, hạn chế trẻ sinh ra bị bệnh. Theo cơ chế di truyền, khi cả vợ và chồng cùng mang gen bệnh thalassemia thì mỗi lần sinh có 25% nguy cơ con bị bệnh, 50% khả năng con mang một gen bệnh và 25% khả năng con bình thường. Việc sàng lọc ra những người có nguy cơ mang gen bệnh thalassemia rất đơn giản bằng xét nghiệm tổng phân tích tế bào máu ngoại vi. Nếu cặp vợ chồng có nguy cơ mang gen bệnh thì cần chẩn đoán sớm cho thai xem có mắc bệnh thalassemia thể nặng không, nhằm giúp ngừng thai nghén sớm, giúp giảm sinh ra những trẻ mắc bệnh thalassemia thể nặng.
Mục đích của sàng lọc và chẩn đoán trước sinh bệnh thalassemia là nhằm chẩn đoán được kiểu gen của thai ở tuần thai sớm nhất có thể. Quy trình chẩn đoán trước sinh bao gồm các bước: 1) Sàng lọc sớm để phát hiện các cặp vợ chồng có nguy cơ sinh con mắc bệnh thalassemia, 2) xác định đột biến gây bệnh của các cặp vợ chồng này, 3) Lấy được các chất liệu di truyền từ thai nhi một cách an toàn và nhanh nhất để chẩn đoán, 4) Xác định kiểu gen của thai bằng phân tích ADN thai dựa trên kiểu đột biến của bố và mẹ. Dưới đây là sơ đồ tư vấn di truyền bệnh thalassemia:
+ Tỷ lệ Hb Bart: 0,25%.
+ Tỷ lệ HbH: 0,79%.
+ Tỷ lệ Beta thalassemia và Beta thalassemia/HbE: 0,14%.
Theo một nghiên cứu về thalassemia được thực hiện tại bệnh viện đa khoa tỉnh Quảng Trị năm 2020 cho kết quả như sau: Bệnh nhân chủ yếu là đối tượng trẻ dưới 20 tuổi (54,9%) và không có sự khác biệt nhiều về giới, bệnh gặp ở cả người Kinh và ngươi Bru Vân Kiều, bệnh nhân vào viện chủ yếu vì tình trạng thiếu máu (62.7%) với biểu hiện da, niêm mạc nhợt nhạt (90,2%), gặp nhiều nhất là β thalasemia (72.5%), ít gặp hơn là α thalasemia (27.5%), mức độ thiếu máu trung bình đến nặng chiếm đa số (68,7%), quá tải sắt (45,1%), tổng đơn vị máu truyền trong năm 282 đơn vị/ 51 bệnh, trung bình 5.5 đơn vị /bệnh /năm.
3. Một số giải pháp
Theo kinh nghiệm của các nước trên thế giới, thalassemia là bệnh có thể chữa được, phòng được, tiến tới chấm dứt tình trạng phù thai do Thalassemia, tình trạng trẻ sinh ra bị bệnh và giảm dần tỷ lệ di truyền gen bệnh trong cộng đồng. Mặt khác, qua thực tiễn cho thấy, chi phí đầu tư cho phòng bệnh nhỏ hơn rất nhiều so với chi phí điều trị và hiệu quả mang lại rất cao.
Có ba giải pháp chính sau:
- Thứ nhất là chẩn đoán và điều trị sớm bệnh thalassemia giúp nâng cao chất lượng cuộc sống của người bệnh.
- Thứ hai là tư vấn tiền hôn nhân, giúp cho người dân biết mình có mang gen bệnh không và có nguy cơ sinh con bị bệnh thalassemia thể nặng không, nhưng không ngăn cản được việc kết hôn.
- Thứ ba là sàng lọc và chẩn đoán trước sinh, giúp chẩn đoán sớm thai bị bệnh thalassemia thể nặng ở tuổi thai nhỏ để tư vấn cho gia đình có thể ngừng thai nghén, giúp cho gia đình và xã hội giảm những gánh nặng chăm sóc và điều trị những người bệnh thalassemia thể nặng.
Trong đó tư vấn tiền hôn nhân, sàng lọc và chẩn đoán trước sinh đóng vai trò quan trọng trong việc dự phòng ngay từ đầu, hạn chế trẻ sinh ra bị bệnh. Theo cơ chế di truyền, khi cả vợ và chồng cùng mang gen bệnh thalassemia thì mỗi lần sinh có 25% nguy cơ con bị bệnh, 50% khả năng con mang một gen bệnh và 25% khả năng con bình thường. Việc sàng lọc ra những người có nguy cơ mang gen bệnh thalassemia rất đơn giản bằng xét nghiệm tổng phân tích tế bào máu ngoại vi. Nếu cặp vợ chồng có nguy cơ mang gen bệnh thì cần chẩn đoán sớm cho thai xem có mắc bệnh thalassemia thể nặng không, nhằm giúp ngừng thai nghén sớm, giúp giảm sinh ra những trẻ mắc bệnh thalassemia thể nặng.
Mục đích của sàng lọc và chẩn đoán trước sinh bệnh thalassemia là nhằm chẩn đoán được kiểu gen của thai ở tuần thai sớm nhất có thể. Quy trình chẩn đoán trước sinh bao gồm các bước: 1) Sàng lọc sớm để phát hiện các cặp vợ chồng có nguy cơ sinh con mắc bệnh thalassemia, 2) xác định đột biến gây bệnh của các cặp vợ chồng này, 3) Lấy được các chất liệu di truyền từ thai nhi một cách an toàn và nhanh nhất để chẩn đoán, 4) Xác định kiểu gen của thai bằng phân tích ADN thai dựa trên kiểu đột biến của bố và mẹ. Dưới đây là sơ đồ tư vấn di truyền bệnh thalassemia:
Tại bệnh viện đa khoa tỉnh Quảng Trị việc chẩn đoán và điều trị thalassemia đã được thực hiện trong nhiều năm qua và mang lại nhiều thành quả tích cực. Từ năm 2019, Bệnh viện đã triển khai dịch vụ điện di huyết sắc tố để chẩn đoán thalassemia. Vấn đề điều trị thalassemia luôn được các khoa lâm sàng quan tâm và thực hiện tốt theo đúng phác đồ hướng dẫn của Bộ y tế. Khoa Huyết học-Truyền máu phối hợp tốt với các khoa lâm sàng trong công tác dự trù và cung cấp máu kịp thời và đầy đủ để truyền cho bệnh nhân khi có chỉ định. Chính những điều trên đã góp phần nâng cao chất lượng cuộc sống cũng như hạn chế những biến chứng bất lợi cho bệnh nhân thalassemia khi điều trị tại Bệnh viện.
4. Kết luận
Bệnh nhân thalassemia phải điều trị bằng truyền máu và thải sắt suốt đời với gánh nặng về suy giảm thể chất, áp lực tâm lý, làm ảnh hưởng đến chất lượng cuộc sống của chính người bệnh và vô hình chung trở thành gánh nặng về kinh tế cho gia đình, cho xã hội khi phải gánh chịu chi phí điều trị bệnh, suy giảm hoặc mất nhân công lao động do phải chăm sóc người thân đi viện liên tục. Điều này ảnh hưởng rất lớn đến chất lượng dân số và việc cải thiện giống nòi sẽ rất khó khăn khi tình trạng mang gen Thalassemia cao trong cộng đồng và tỷ lệ trẻ sinh ra bị bệnh thalassemia ngày một tăng do chưa khống chế được nguồn gen bệnh lây lan âm ỉ trong cộng đồng, đặc biệt ở các dân tộc ít người và rất ít người. Như vậy, có thể nói: “Ở Việt Nam, “quả bom nguyên tử Thalassemia” đã nổ, nhưng chúng ta không nghe được tiếng nổ của nó, mặc dù hậu quả gây ra đã rất nghiêm trọng rồi!”.
Để ngăn chặn bệnh thalassemia thực sự cần những chương trình hành động cụ thể và sự chung sức, đồng lòng của cả hệ thống xã hội, từ y tế, giáo dục, dân số, các tổ chức chính trị, xã hội… và của cả cộng đồng đúng như thông điệp của Ngày thalassemia Thế giới (8/5) “Nhận thức–Chia sẻ–Quan tâm: Chung tay cùng cộng đồng quốc tế để nâng cao nhận thức về bệnh thalassemia”.
Một số đường link: https://www.youtube.com/watch?v=3NMnuFmtGh8&t=10s
https://www.youtube.com/watch?v=6InZAL8FSiI&t=56s
https://www.facebook.com/100070682894471/posts/176721138027315/
TÀI LIỆU THAM KHẢO
1. Bộ y tế (2015). Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học. Nhà Xuất Bản Y học, 233-237.
2. Bernadette Modella & Matthew Darlisona (2008). Global epidemiology of haemoglobin disorders and derived service indicators, Bulletin of the World Health Organization 2008; 86: 480 – 487.
3. Thalassemia Internation Federation annual report (2013).
4. Nguyễn Anh Trí, Bạch Quốc Khánh và cs (2020). Nghiên cứu đặc điểm dịch tễ gen bệnh thalassemia/huyết sắc tố tại Việt Nam. Đề tài cơ sở Viện Huyết học -Truyền máu TW.
5. Nguyễn Thị Thu Hà và cs (2021): Tổng quan thalassemia, thực trạng, nguy cơ và giảm pháp kiểm soát bệnh thalassemia ở Việt Nam, Tạp chí Y học Việt Nam, tập 502 tháng 5/2021.
4. Kết luận
Bệnh nhân thalassemia phải điều trị bằng truyền máu và thải sắt suốt đời với gánh nặng về suy giảm thể chất, áp lực tâm lý, làm ảnh hưởng đến chất lượng cuộc sống của chính người bệnh và vô hình chung trở thành gánh nặng về kinh tế cho gia đình, cho xã hội khi phải gánh chịu chi phí điều trị bệnh, suy giảm hoặc mất nhân công lao động do phải chăm sóc người thân đi viện liên tục. Điều này ảnh hưởng rất lớn đến chất lượng dân số và việc cải thiện giống nòi sẽ rất khó khăn khi tình trạng mang gen Thalassemia cao trong cộng đồng và tỷ lệ trẻ sinh ra bị bệnh thalassemia ngày một tăng do chưa khống chế được nguồn gen bệnh lây lan âm ỉ trong cộng đồng, đặc biệt ở các dân tộc ít người và rất ít người. Như vậy, có thể nói: “Ở Việt Nam, “quả bom nguyên tử Thalassemia” đã nổ, nhưng chúng ta không nghe được tiếng nổ của nó, mặc dù hậu quả gây ra đã rất nghiêm trọng rồi!”.
Để ngăn chặn bệnh thalassemia thực sự cần những chương trình hành động cụ thể và sự chung sức, đồng lòng của cả hệ thống xã hội, từ y tế, giáo dục, dân số, các tổ chức chính trị, xã hội… và của cả cộng đồng đúng như thông điệp của Ngày thalassemia Thế giới (8/5) “Nhận thức–Chia sẻ–Quan tâm: Chung tay cùng cộng đồng quốc tế để nâng cao nhận thức về bệnh thalassemia”.
Một số đường link: https://www.youtube.com/watch?v=3NMnuFmtGh8&t=10s
https://www.youtube.com/watch?v=6InZAL8FSiI&t=56s
https://www.facebook.com/100070682894471/posts/176721138027315/
TÀI LIỆU THAM KHẢO
1. Bộ y tế (2015). Hướng dẫn chẩn đoán và điều trị một số bệnh lý huyết học. Nhà Xuất Bản Y học, 233-237.
2. Bernadette Modella & Matthew Darlisona (2008). Global epidemiology of haemoglobin disorders and derived service indicators, Bulletin of the World Health Organization 2008; 86: 480 – 487.
3. Thalassemia Internation Federation annual report (2013).
4. Nguyễn Anh Trí, Bạch Quốc Khánh và cs (2020). Nghiên cứu đặc điểm dịch tễ gen bệnh thalassemia/huyết sắc tố tại Việt Nam. Đề tài cơ sở Viện Huyết học -Truyền máu TW.
5. Nguyễn Thị Thu Hà và cs (2021): Tổng quan thalassemia, thực trạng, nguy cơ và giảm pháp kiểm soát bệnh thalassemia ở Việt Nam, Tạp chí Y học Việt Nam, tập 502 tháng 5/2021.
Tác giả: BS. Bùi Ngọc Hoàng, Khoa Huyết học-Truyền máu
Ý kiến bạn đọc
Bạn cần đăng nhập với tư cách là Thành viên chính thức để có thể bình luận
Những tin mới hơn
Những tin cũ hơn
Kiến thức y khoa
Góc tri ân
Album ảnh bệnh viện
-
 Tọa đàm ngày Quốc tế phụ nữ 8.3.2019
Tọa đàm ngày Quốc tế phụ nữ 8.3.2019
-
 Thành lập đơn vị đột quỵ - thầy thuốc ưu tú
Thành lập đơn vị đột quỵ - thầy thuốc ưu tú
thành lập đơn vị đột quỵ - thầy thuốc ưu tú
-
 Ngày thầy thuốc Việt Nam 27.2.2018
Ngày thầy thuốc Việt Nam 27.2.2018
ngày thầy thuốc Việt Nam 27.2.2018
-
 Hội thi quy tắc ứng xử 2018
Hội thi quy tắc ứng xử 2018
hội thi quy tắc ứng xử 2018
-
 Hội nghị khoa học tiết niệu 12.4.2019
Hội nghị khoa học tiết niệu 12.4.2019
Bạn đã không sử dụng Site, Bấm vào đây để duy trì trạng thái đăng nhập. Thời gian chờ: 60 giây
Gửi phản hồi