Báo cáo nhân một trường hợp bệnh tạo xương bất toàn (Osteogenesis Imperfecta)
Thứ năm - 30/09/2021 10:19
Báo cáo nhân một trường hợp bệnh tạo xương bất toàn (Osteogenesis Imperfecta)
1. Đặt vấn đề
Bệnh tạo xương bất toàn (Osteogenesis Imperfecta) còn gọi là bệnh xương thủy tinh hay bệnh giòn xương. Trường hợp bệnh đầu tiên được mô tả bởi Lobstein vào năm 1833. Hơn 90% bệnh tạo xương bất toàn là di truyền trội trên nhiễm sắc thể thường. Bệnh bao gồm nhiều thể lâm sàng và có đặc điểm di truyền khác nhau với tần suất mắc bệnh 1/20000.
Nguyên nhân của bệnh là do đột biến gen tổng hợp collagen typ 1 dẫn đến thiếu hụt hoặc bất thường cấu trúc của phân tử collagen typ 1 gây nên giòn xương, giảm khối lượng xương và bất thường các mô liên kết. Người mắc bệnh tạo xương bất toàn dễ gãy xương tự nhiên kèm theo các bất thường ở tổ chức khác như: củng mạc mắt màu xanh, yếu cơ, tạo răng bất toàn, tăng vận động các khớp, giảm thính lực (chiếm 50-65% bệnh nhân tạo xương bất toàn). Bệnh tạo xương bất toàn gây đau đớn, tàn phế suốt đời cho trẻ cả về mặt thể chất lẫn tâm thần với thể bệnh nhẹ và tỷ lệ tử vong cao với thể bệnh nặng.
Hiện nay, kỹ thuật phân tích gen là tiền đề quan trọng giúp chẩn đoán trước sinh đối với các đối tượng có nguy cơ cao sinh con bị bệnh tạo xương bất toàn để đưa ra những tư vấn di truyền giúp ngăn ngừa và giảm tỷ lệ mắc bệnh.
Bài viết này chúng tôi báo cáo một trường hợp bệnh tạo xương bất toàn được phát hiện, chẩn đoán và đang điều trị tại Khoa Nhi thời gian vừa qua. 2. Báo cáo ca bệnh
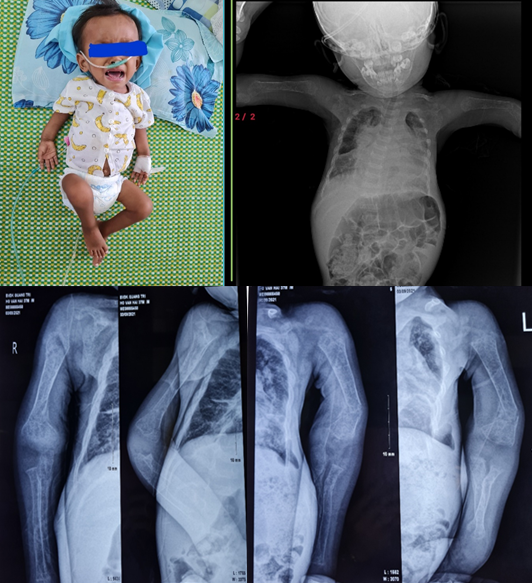
Bệnh nhi nam 37 tháng nhập viện vì sốt, ho, khó thở. Tiền sử trẻ sinh non 36 tuần - 2,5Kg, chậm tăng cân từ 5 tháng tuổi, 7 tháng biết lật, 9 tháng biết bò, chưa đứng vững, chưa biết đi, nói được từng từ một, chưa đi khám và điều trị trước đây. Tiền sử gia đình không ghi nhận bất thường. Ghi nhận lúc vào khoa:Trẻ tinh, suy dinh dưỡng nặng thể teo đét, da môi hồng nhạt, SpO2 96%, nhiệt độ: 38,5 độ C, nhịp thở 50 lần/phút, cân nặng 6kg. Trẻ van đau nhức xương toàn thân thường xuyên, hai tay hai chân biến dạng, gập góc, teo cơ tứ chi. Hạn chế cử động tay, chân. Hàm răng cùn mòn, trắng đục, không có răng hàm trên. Củng mạc mắt màu xanh. Ho nhiều, thở đều, gắng sức. Phổi nghe ran ẩm hai bên Nhịp tim đều, rõ, không nghe tiếng thổi Trẻ được chẩn đoán: Viêm phổi/Suy dinh dưỡng nặng/TD Tạo xương bất toàn Được tiến hành làm các xét nghiệm, chụp phim Xquang Kết quả: Xquang ngực: Tổn thương kẽ lan tỏa hai phổi. Bóng tim lớn, đậm rốn phổi hai bên. Xquang xương cánh tay: Loãng xương lan tỏa xương cánh tay hai bên, gãy cũ kèm biến dạng xương cẳng tay hai bên. Khuyết các điềm cốt hóa khớp khuỷu trái và phải.
Hình ảnh bệnh nhân Hồ Văn H. 37 tháng
Trẻ được điều trị viêm phổi với kháng sinh, dinh dưỡng tĩnh mạch kèm chế độ ăn đảm bảo dinh dưỡng cho trẻ suy dinh dưỡng nặng, bù điện giải, giảm đau. Sau 2 tuần, tình trạng viêm phổi cải thiện, tiến hành điều trị thuốc Biphosphonate (Acid Zoledronic) theo phác đồ. Hiện tại, trẻ tỉnh, giảm tình trạng đau nhức xương, tay chân có cử động được, tình trạng viêm phổi cải thiện. 3. Tổng quan về bệnh tạo xương bất toàn Chẩn đoán bệnh tạo xương bất toàn chủ yếu dựa vào triệu chứng lâm sàng, Xquang, tiền sử gãy xương, tiền sử gia đình. 3.1. Triệu chứng lâm sàng: Lâm sàng phụ thuộc vào từng typ của bệnh tạo xương bất toàn với các biểu hiện:
- Loãng xương, gãy xương tự phát và/hoặc tái phát
- Củng mạc mắt màu xanh
- Tạo răng bất toàn
- Giảm thính lực
- Người thấp, bé
- Khớp lỏng lẻo
- Yếu cơ, mỏi cơ
- Khuôn mặt hình tam giác, lồng ngực hình thùng
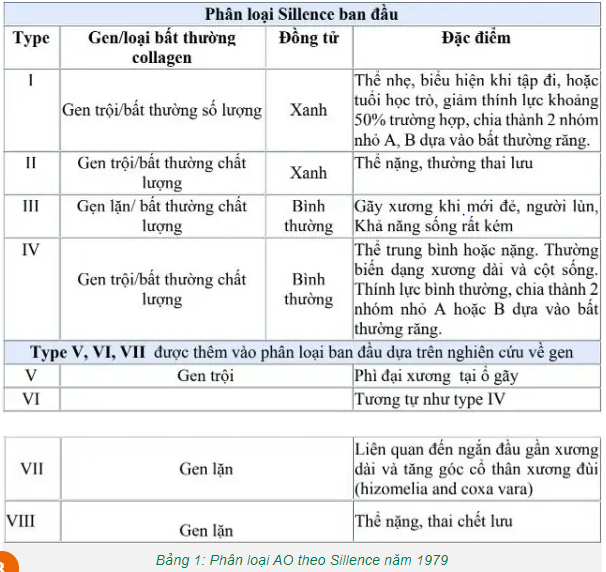
Sillence và cộng sự (1979) đã chia bệnh tạo xương bất toàn thành bốn typ
- Typ I: là thể nhẹ nhất và hay gặp nhất. Người bệnh có tầm vóc bình thường hoặc tương đối bình thường, có biểu hiện yếu cơ, cột sống có thể bị cong, củng mạc mắt có thể có màu xanh hay màu tím, gãy xương thường xảy ra trước tuổi dậy thì. - Typ II: là thể nặng nhất. Bệnh nhân thường chết ngay sau khi sinh hoặc chỉ sống được một thời gian ngắn do rối loạn chức năng hô hấp (thiểu sản phổi, gãy xương sườn), người bệnh có vóc dáng nhỏ, gãy nhiều xương. - Typ III: là thể tương đối nặng, trẻ thường sinh ra đã có gãy xương. Củng mắt thường quá trắng hoặc có màu xám, màu xanh, giảm chức năng hô hấp, giảm thính lực và bất thường về răng. - Typ IV: là thể trung gian giữa typ I và III. Các biến dạng xương ở mức nhẹ đến trung bình. Xếp theo thứ tự từ nhẹ đến nặng sẽ là typ I, typ IV, typ III, typ II.
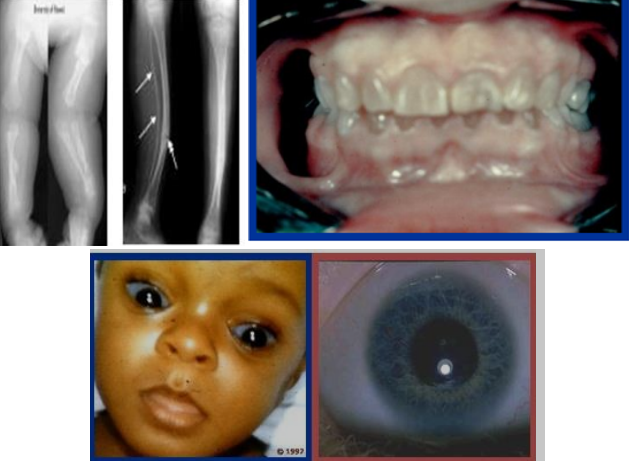
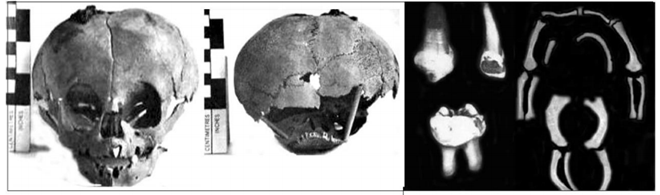
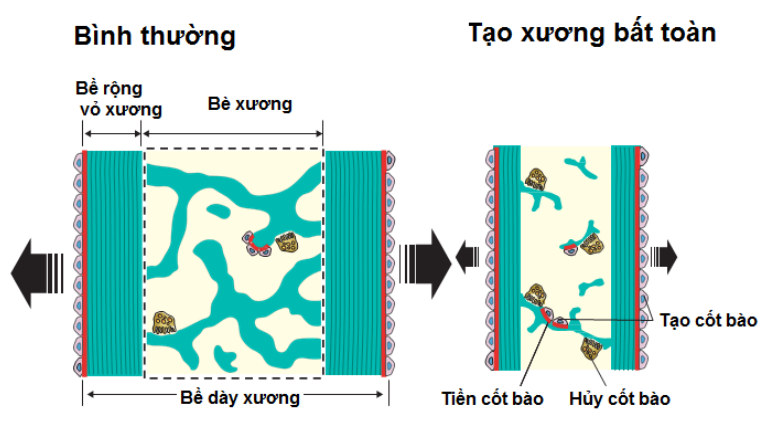
Một số nghiên cứu mới đã phân loại bệnh tạo xương bất toàn thành 16 typ. Thể bệnh tạo xương bất toàn được phân loại tùy theo mức độ nghiêm trọng của bệnh. 3.2. Cận lâm sàng 3.2.1. Xquang Xét nghiệm cận lâm sàng có giá trị trong chẩn đoán bệnh tạo xương bất toàn là chụp Xquang đơn thuần. Biểu hiện trên Xquang thông thường của bệnh được phân loại theo các typ. Typ I: Xương sọ Tam O’Shanter: sọ phẳng ở trục thẳng đứng và rộng theo chiều ngang. Vỏ xương mỏng, xương dài gãy và biến dạng, giảm độ đặc của xương, đốt sống dẹt, mất chất vôi. Typ II: Xương sườn hình chuỗi hạt, xương bè ngang, gãy và biến dạng xương, giảm độ đặc của xương, dẹt đốt sống. Typ III: nang hành xương, xương bình thường hoặc bè ở giai đoạn sớm, mỏng ở giai đoạn muộn, gãy và biến dạng xương, giảm độ đặc của xương. Typ IV: vỏ xương mỏng, gãy và biến dạng xương, dẹt đốt sống
Phần còn lại của xương một trẻ bị bệnh tạo xương bất toàn với hình ảnh xương sọ Tam O’Shanter, tạo răng bất toàn, xương mỏng và biến dạng
3.2.2. Sinh thiết xương Là xét nghiệm mô bệnh học từ mẫu bệnh phẩm sinh thiết xương dùng để chẩn đoán và theo dõi điều trị. Xương của bệnh nhân tạp xương bất toàn có kích cỡ nhỏ hơn bình thường, giảm số lượng bè xương và các bè xương mỏng hơn bình thường.
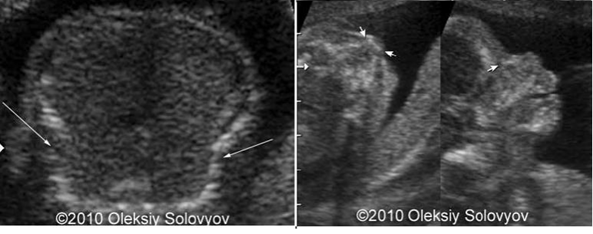
3.2.3. Xét nghiệm di truyền phân tử Phân tích các procolagen loại I từ các nguyên bào sợi nuôi cấy (từ sinh thiết da) hoặc phân tích gen COL1A1 và COL1A2 có thể được sử dụng khi chẩn đoán lâm sàng không rõ ràng. Nuôi cấy nguyên bào sợi từ da bệnh nhân để phân tích xác định cấu trúc và chất lượng của Collagen typ I (độ nhạy 87% đối với thể trung bình nhẹ và 98% với thể nặng). 3.2.4. Các xét nghiệm máu khác Công thức máu, điện giải đồ, phospho, phosphatase kiềm, chức năng thận thường trong giới hạn bình thường. Xét nghiệm nước tiểu có canxi niệu tăng hoặc bình thường. Siêu âm trước sinh có thể chẩn đoán chính xác bệnh đến 89%. Việc chẩn đoán có thể thực hiện sớm nhất ở tuần 17 với typ I và tuần 13 với typ II. Bệnh typ III hoặc IV có thể có hoặc không có gãy xương, biến dạng xương hoặc giảm khoáng hóa xương, nếu có thì sự biến đổi chi nhìn thấy được ở 3 tháng cuối thai kỳ.
Hình ảnh siêu âm thai ở 20 tuần (Mũi tên cho thấy gãy xương sườn, giảm khoáng ở xương mũi)
4. Điều trị
Hiện chưa có phương pháp điều trị dứt điểm, liệu pháp gen là phương pháp đang được nghiên cứu và là hy vọng trong điều trị tận gốc bệnh, các phương pháp điều trị hiện nay nhằm mục đích cải thiện chất lượng sống cho người bệnh, hạn chế tối đa gãy xương và biến chứng của gãy xương, một số phương pháp đang được áp dụng bao gồm: - Sử dụng hormone tăng trưởng giúp trẻ đáp ứng nhu cầu tăng trưởng ở nhóm I và IV. - Điều trị Bisphophosnates và teriparatide nhằm tăng mật độ xương, giảm đau xương và giảm nguy cơ gãy xương. - Điều trị phẫu thuật trong trường hợp gãy xương, biến dạng chi, vẹo cột sống, chèn ép nền sọ. - Điều trị nha khoa. - Vật lý trị liệu, sử dụng dụng cụ hỗ trợ, điều trị nâng đỡ, dinh dưỡng. - Cấy ốc tai trong trường hợp bé khiếm thính. - Thuốc ức chế sinh học, hormone (GH, estrogen), ghép tế bào gốc, điều trị gen đang được áp dụng và đánh giá kết quả. TÀI LIỆU THAM KHẢO
Bùi Thị Hồng Châu, Trần Vân Khánh, Hồ Cẩm Tú, Trần Huy Thịnh, Tạ Văn Thành (2013), “Đột biến gen COL1A1 trên bệnh nhân tạo xương bất toàn”, Tạp chí Y học thành phố Hồ Chí Minh, 17(1), tr 20-24.
Antoniazzi F, Mottes M, Fraschini P, et al (2000), Osteogenesis Imperfecta: Practical treatment guidelines, Paediatr Drugs, 2000, 2: 465.
John F Beary, III, MD, (2021), Osteogenesis Imperfecta: An overview, Uptodate.
Kwang-Soo Lee, Hae-Ryong Song, Tae-Joon Cho, Sung-Chul Jung, and Soo Kyung Koo (2006). Mutational Spectrum of Type I Collagen Genes in Korean Patients with Osteogenesis Imperfecta. Human Mutation, 27(6).
Marom R, Rabenhorst BM, Morello R (2020), osteogenesis imperfecta: An update on clinical features and therapies, Eur J Endocrinol, 183:R95.
Rauch F and Glorieux FH (2004). “Osteogenesis imperfecta”. Lancet, 363, pp 1377-1385.
Stoll C., Dott B., Roth M.P., Alembik Y. (1989). Birth prevalence rates of skeletal dysplasias. Clin Genet, 35(2), 88-92.
Sillence D.O, Senn A, Danks D.M (1979). “Genetic heretogeneity in Osteogenesis Imperfecta”. J.Med.Genet, 16, pp101-106.
Van Dijk F.S. (2013). Clinical utility gene card: for osteogenesis imperfecta. European Journal of Human Genetics, 21, 6.
 Bệnh viện đa khoa tỉnh Quảng Trị | Bệnh viện tỉnh Quảng Trị | BVĐK Quảng Trị
Bệnh viện đa khoa tỉnh Quảng Trị | Bệnh viện tỉnh Quảng Trị | BVĐK Quảng Trị

 Tọa đàm ngày Quốc tế phụ nữ 8.3.2019
Tọa đàm ngày Quốc tế phụ nữ 8.3.2019
 Thành lập đơn vị đột quỵ - thầy thuốc ưu tú
Thành lập đơn vị đột quỵ - thầy thuốc ưu tú
 Ngày thầy thuốc Việt Nam 27.2.2018
Ngày thầy thuốc Việt Nam 27.2.2018
 Hội thi quy tắc ứng xử 2018
Hội thi quy tắc ứng xử 2018
 Hội nghị khoa học tiết niệu 12.4.2019
Hội nghị khoa học tiết niệu 12.4.2019